Technology Transfer Network
Emission Measurement Center Spectral Database
Coal-Fired Fluidized Bed Boiler Emission Test
|
TEXAS-NEW MEXICO POWER COMPANY UNIT 2 HAMMOND, TEXAS EPA Contract No. 68D20163 Work Assignment No. I-34 Prepared by: Research Division Entropy, Inc. Post Office Box 12291 Research Triangle Park, North Carolina 27709 Prepared for: Lori Lay U. S. Environmental Protection Agency Emissions Measurement Branch Research Triangle Park, North Carolina 27711 June 15, 1994 DISCLAIMER This document was prepared by Entropy, Inc. under EPA Contract No. 68D20163, Work Assignment No. I-34. This document has been reviewed by the U.S. Environmental Protection Agency (EPA). The opinions, conclusions, and recommendations expressed herein are those of the authors, and do not necessarily represent those of EPA. Mention of specific trade names or products within this report does not constitute endorsement by EPA or Entropy, Inc. TABLE OF CONTENTS 1.0 INTRODUCTION 1.1 BACKGROUND 1.2 DESCRIPTION OF THE PROJECT 1.3 PROJECT ORGANIZATION 2.0 PROCESS DESCRIPTION AND SAMPLE POINT LOCATIONS 2.1 PROCESS DESCRIPTION 2.2 CONTROL EQUIPMENT DESCRIPTION 2.3 SAMPLE POINT LOCATIONS 3.0 SUMMARY AND DISCUSSION OF RESULTS 3.1 OBJECTIVES AND TEST MATRIX 3.2 FIELD TEST CHANGES AND PROBLEMS 3.3 SUMMARY OF RESULTS 4.0 SAMPLING AND ANALYTICAL PROCEDURES 4.1 EXTRACTIVE SYSTEM FOR DIRECT GAS PHASE ANALYSIS 4.2 SAMPLE CONCENTRATION 4.3 CONTINUOUS EMISSIONS MONITORING 4.4 FLOW DETERMINATIONS 4.5 PROCESS OBSERVATIONS 4.6 ANALYTICAL PROCEDURES 5.0 QUALITY ASSURANCE/QUALITY CONTROL ACTIVITIES 5.1 QC PROCEDURES FOR MANUAL FLUE GAS TEST METHODS 5.2 QC PROCEDURES FOR INSTRUMENTAL METHODS 5.3 QA/QC CHECKS FOR DATA REDUCTION, VALIDATION, AND REPORTING 5.4 CORRECTIVE ACTIONS 6.0 CONCLUSIONS 7.0 REFERENCES APPENDICES NOTE: Appendices A-D are not available 1.0 INTRODUCTIONThe U. S. Environmental Protection Agency (EPA) Office of Air Quality Planning and Standards (OAQPS), Industrial Studies Branch (ISB), and Emission Measurement Branch (EMB) directed Entropy, Inc. to conduct an emission test at Texas-New Mexico Power Company's (TNP) Unit 2 fluidized bed coal-fired boiler, in Hammond, Texas. The test was conducted from May 10 to May 13, 1993. The purpose of this test was to identify which hazardous air pollutants (HAPs) listed in the Clean Air Act Amendments of 1990 are emitted from this source. The measurement method used Fourier transform infrared (FTIR) technology, which had been developed for detecting and quantifying many organic HAPs in a flue gas stream. Besides developing emission factors (for this source category), the data will be included in an EPA report to Congress. Before this test program, Entropy conducted screening tests using the FTIR method at facilities representing several source categories, including a coal-fired boiler. These screening tests were part of the FTIR Method Development project sponsored by EPA to evaluate the performance and suitability of FTIR spectrometry for HAP emission measurements. The screening tests helped determine sampling and analytical limitations, provided qualitative information on emission stream composition, and allowed estimation of the mass emission rates for a number of HAPs detected at many process locations. The evaluation demonstrated that gas phase analysis using FTIR can detect and quantify many HAPs at concentrations in the low part per million (ppm) range and higher, and a sample concentration technique was able to detect HAPs at sub-ppm levels. Following the screening tests, Entropy conducted a field validation study at a coal-fired steam generation facility to assess the effectiveness of the FTIR method for measuring HAPs, on a compound by compound basis. The flue gas stream was spiked with HAPs at known concentrations so that calculated concentrations, provided by the FTIR analysis, could be compared with actual concentrations in the gas stream. The analyte spiking procedures of EPA Method 301 were adapted for experiments with 47 HAPs. The analytical procedures of Method 301 were used to evaluate the accuracy and precision of the results. Separate procedures were performed to validate a direct gas phase analysis technique and a sample concentration technique of the FTIR method. A complete report, describing the results of the field validation test, has been submitted to EPA.[1] This report was prepared by Entropy, Inc. under EPA Contract No. 68D20163, Work Assignment No. I-34. The field test was performed under EPA Contract No. 68D20163 Work Assignment No. 4. Research Triangle Institute (RTI) provided the process information presented in Sections 2.1 and 3.3.3. 1.2 DESCRIPTION OF THE PROJECT The FTIR-based method uses two different sampling techniques: (1) direct analysis of the extracted gas stream (hereafter referred to as the gas phase technique or gas phase analysis) and (2) sample concentration followed by thermal desorption. Gas phase analysis involves extracting gas from the sample point location and transporting the gas through sample lines to a mobile laboratory where sample conditioning and FTIR analyses are performed. The sample concentration system employs 10 g of Tenax® sorbent, which can remove organic compounds from a flue gas stream. Organic compounds adsorbed by Tenax® are then thermally desorbed into the smaller volume of the FTIR absorption cell; this technique allows detection of some compounds down to the ppb level in the original sample. For this test, approximately 850 dry liters of flue gas were sampled during each run using sample concentration. Section 4.0 describes the sampling systems. Entropy operated a mobile laboratory (FTIR truck) containing the instrumentation and sampling equipment. The truck was driven to the site at TNP and parked next to the Unit One baghouse below the inlet sampling location. Four test runs were performed over a three-day period. Entropy tested the exhaust gases from three locations on TNP Unit 2:
The outlet of the air preheater upstream of the baghouse.
The outlet of the baghouse in the breaching upstream of the stack.
The stack.
Four 4-hour sample concentration runs were performed at the air preheater outlet and the stack. Gas phase runs were performed concurrently with sample concentration at the air preheater outlet and the baghouse outlet. Flow measurements were performed at the air preheater outlet and at the stack. Section 2.1 contains the sampling location descriptions and Section 3.1 contains the test schedule. Direct gas phase analysis was used to measure carbon monoxide (CO), carbon dioxide (CO2), sulfur dioxide (SO2), nitrogen oxides (NOx), and ppm levels of other species. EPA instrumental test methods were used to provide concentrations of CO, CO2, O2, and hydrocarbons (HC). The sample concentration technique was used to measure HAPs at ppb levels. The test program was funded and administered by the Industrial Studies Branch (ISB) and the Emissions Measurement Branch (EMB) of the U.S. EPA. A representative from Research Triangle Institute (RTI) collected process data. The following list presents the organizations and personnel involved in coordinating and performing this project.
TNP Corporate Contact: Dr. Larry L. Sandburg (817) 746-7604
TNP Plant Contact: Mr. Zan Shafer (817) 746-7604
EMB Work Assignment Ms. Lori Lay (919) 541-4825
Managers: Mr. Dennis Holzschuh (919) 541-5239
Industrial Studies Branch Mr. Kenneth Durkee (919) 541-5425
(ISB) Contacts: Mr. William Maxwell (919) 541-5430
Entropy Project Manager: Dr. Thomas Geyer (919) 781-3551
Entropy Test Personnel: Dr. Laura Kinner
Mr. Scott Shanklin
Ms. Lisa Grosshandler
Mr. Greg Blanschan
Mr. Mike Worthy
Mr. Rick Straughsbaugh
Dr. Ed Potts
Dr. Craig Stone
RTI Representative: Mr. Jeffrey Cole (919) 990-8606
2.0 PROCESS DESCRIPTION AND SAMPLE POINT LOCATIONSThe process information given in this section was supplied by TNP personnel. A process air flow diagram is presented in Figure 2-1. TNP One Unit Two is a base-loaded unit that normally operates 24 hours a day, 7 days a week, except for an annual 2- to 3-week planned outage for maintenance. The primary fuel source for Unit Two is the Walnut Creek Mining Company. This drag-line-equipped lignite mine is located adjacent to the plant. Trucks haul the lignite from the mine to a covered coal storage building. After being crushed to approximately 3/8-inch diameter, the coal is carried by a conveyer to one of four holding silos. While being transported to the silos the coal is sampled every 60 seconds by an automatic sampler, which grinds the sampled coal and places it in a sampling container. From the silos, the coal is fed simultaneously, at a controlled flow rate, through four injection systems into the atmospheric circulating fluidized bed (ACFB) combustor. During the test program, Unit Two had an average coal consumption of 113.83 ton/hr. TNP One is also able to use natural gas, petroleum coke, or western coal as alternate fuels. None of these alternate fuels was used during the test period. The limestone used for sulfur dioxide (SO2) control is supplied by Texas Crushed Stone, Inc., of Williamson County, Texas. It is hauled to the plant by train and pulverized before being fed into one of two limestone storage silos. The limestone is introduced into the combustor by an air-fluidized system via four injection points located near the four lignite injection points. During testing, Unit Two had an average limestone injection rate of 6.75 ton/hr. Lignite cannot be used at startup to fuel the ACFB. During startup operations, Unit Two uses natural gas for fuel. To provide a bed material, which is necessary for ACFB operation, limestone is used. This combination of natural-gas-heated fluidizing air and limestone is used to bring the unit up to minimum lignite combustion temperature, at which time the lignite is introduced. Eventually the natural gas is turned off completely. The ACFB combustor produces 1,100,000 lb/hr of steam for a Westinghouse steam turbine generator rated at 160 MWe. The ACFB technology is a process that allows for gas/solids reaction. Combustion in the ACFB takes place in a vertical chamber called the combustor. Inside the chamber, solids (lignite and limestone) are introduced and recirculated until they are calcined (heated to drive off volatile compounds and produce a powdery residue). Normal operating temperature in the combustor is approximately 1,560 degF. The limestone captures the SO2 released from the burning lignite and reacts to form calcium sulfate (gypsum). The bed material in the combustor consists primarily of lignite ash, gypsum, and excess calcined lime. The mean particle size of the bed material is in the range of 50 to 300m. The bed material is fluidized with primary air introduced through a grate at the bottom of the combustor and by the combustion gas generated. The combustion gas flows upward with a relatively high fluidizing velocity. Secondary air is introduced to the combustion process through the sidewalls and bottom of the combustor. Roughly 60 percent of the combustion air is introduced as primary air, and the balance is admitted as secondary air. Combustion takes place in two zones of the combustor: in the primary reducing zone (lower section), and then in the upper section where combustion is completed. Flue gas, resulting from the combustion of the fuel, and entrained solids exit the combustor via gas outlets located in the upper portion of the combustor. Ducts carry flue gas to recycling cyclones designed to remove 99.7 percent of the solids entrained in the gas. Some of the solids separated by the recycling cyclones are collected and returned directly to the combustor. The remainder of the solids pass by the Spiece valves (used to adjust flow) and through fluid bed heat exchangers containing superheat, reheat, and evaporative tube bundles. The cooled solids from these exchangers are then returned to the combustor. From the top of the recycling cyclones, the flue gas enters the extended backpass section, convective backpass section, and the air preheaters on its way to the fabric filter. There are two air preheaters, primary and secondary. The primary and secondary air preheater ducts meet just before the fabric filter (baghouse) entrance. The primary duct is larger than the secondary duct, which requires an air tempering damper on the secondary air duct to keep airflow and temperature even. The fabric filter is a negative pressure design utilizing two induced draft (ID) fans rated at 3,000 hp each. These fans are located on the clean side of the fabric filter. From the fabric filter exit, the flue gas enters a 340-ft stack with a 12-ft exit diameter, then is exhausted into the atmosphere. 2.2 CONTROL EQUIPMENT DESCRIPTION 2.2.1 Nitrogen Oxides (NOx) Control Unit Two is, by design, a low NOx unit because of low combustion temperatures. However, as an added NOx control, secondary air can be used as overfire air to further control NOx. Although operable since the unit began operation, the overfire air system has not been needed for NOx control. 2.2.2 Sulfur Dioxide (SO2) Control As stated above, the limestone captures the SO2 released from the burning lignite and reacts to form calcium sulfate (gypsum). 2.2.3 Particulate Control A fabric filter is used to collect the flyash that exits the air preheaters. The flyash is collected on the inside of tubular filter bags. The system is a shake-deflate fabric filter built by Southwest Public Service Co. (SPS) and uses a total of 3,024 Teflon®-coated fiberglass filter bags (216 per compartment) distributed in 14 compartments ( Figure 2-2). Each bag is 11.5 inches in diameter and 33.67 feet long. The fabric filter is designed to clean 650,000 acfm of flue gas at the air preheater outlet temperature (exact design temperature not available). Design efficiency is 99.78 percent based on an inlet ash loading of 22,000 lb/hr (3.95 grains/ft3) and an outlet ash loading of 48.8 lb/hr (0.03 lb/106 Btu). With all 14 compartments operating (normal operation), the gross air-to-cloth ratio for the fabric filter is 2.12:1. There are two cycles in the fabric filter system: the filtering cycle and the cleaning cycle. The compartment cleaning cycle is initiated every 3 hours or on a high differential pressure across the fabric filter. When the pressure reaches 5 in. w.g., the cleaning sequence is initiated, which involves a compartment-by-compartment isolation and cleaning. This cleaning takes about 60 to 90 seconds per compartment or 14 to 21 minutes for the entire fabric filter. 2.3.1 Air Preheater Outlet (Baghouse Inlet) Separate ducts carrying flue gases exiting the secondary air preheater outlet and the primary air preheater outlet combine upstream of the baghouse (see Figure 2-3). The duct leading from the primary air outlet is 14 ft 9 in wide and 6 ft 11 in deep. The duct leading from the secondary air outlet is 10 ft wide by 6 ft 11 in. Three, evenly spaced, four-inch diameter sampling ports were available on each of these ducts approximately 5 feet upstream of the meeting point of the two streams. The components of the gas stream were expected to be well mixed, and samples were extracted from only the primary air outlet duct. Velocity traverses were made through all ports on both ducts using a 3-dimensional pitot probe. 2.3.2 Baghouse Outlet Before testing, TNP installed a port in the breaching immediately after the baghouse upstream of the induced draft (I.D.) fan and the stack. This location allowed Entropy to perform gas phase test runs concurrently at the baghouse inlet and outlet locations (Section 4.1). No flow measurements were taken at the baghouse outlet. With the approval of EPA, volumetric flow at this location was assumed to be the same as at the stack. 2.3.3 Exhaust Stack A 5 ft wide platform at the 245 ft level of the stack was accessible by an elevator (Figure 2-4). Two 6-inch diameter ports were reached through access panels in the concrete shell that surrounds the entire stack. Continuous emission monitors, for SO2, and NOx, had been installed by TNP at the same level of the stack. The analyzers were operating during the time of the test and TNP offered to make these data available. The sample concentration probe was inserted in one of the ports and the sample train was set up on the platform. Gas phase measurements were not performed at the stack due to the length of line required to reach this location. 3.0 SUMMARY AND DISCUSSION OF RESULTS3.1 OBJECTIVES AND TEST MATRIX The purpose of the test program was to obtain information that will enable EPA to develop emission factors (for as many HAPs as possible) which will apply to electric utilities employing coal-fired boilers. EPA also plans to use these results in a report for Congress. The specific objectives were:
Measure HAP emissions (using FTIR spectrometry) in two
concentration ranges, 1 ppm and higher using gas phase analysis,
and below 1 ppm using sample concentration/thermal desorption.
Determine maximum possible concentrations for undetected HAPs based
on detection limits of instrumental configuration and limitations
imposed by composition of flue gas matrix.
Measure O2, CO2, CO, and hydrocarbons using gas analyzers.
Perform simultaneous test runs at inlet/outlet locations and
analyze data to assess effect (if any) of control device (baghouse)
on HAP (and other pollutant) emissions.
Employ 3-dimensional probe to develop flow profile of air preheater
outlet locations and determine volumetric flow rates.
Obtain process information from TNP. This information includes the
rate of power production and operating parameters of the control
devices during the test periods.
Correlate test results with process data.
Table 3-1 presents the test schedule that was followed at TNP. 3.2 FIELD TEST CHANGES AND PROBLEMS The thermally desorbed samples from Run 1 contained high levels of water vapor. The spectra were collected and saved and have been analyzed, but their interpretation is complicated by larger than usual infrared absorbance from water vapor. One advantage of sample concentration/thermal desorption is that spectral interferences from water and CO2 can be significantly decreased. This advantage, which permits analysis in spectral regions that are normally unavailable in gas phase samples, was lost in samples from Run 1. Because the concentrated samples from Run 1 were very wet, a fourth run was added to the schedule in order to ensure that good quality sample concentration data were obtained for at least three runs. Runs 3 and 4 were both performed on May 13, so the revised test schedule could be completed in the originally planned time. Sample concentration Run 3 went from 9:20 A.M. to 1:20 P.M. Sample concentration Run 4 went from 2:00 to 6:00 P.M. Gas phase data collection and CEM analysis went from noon to 4:15 P.M. Direct gas phase analysis was stopped before the end of Run 4 because sufficient data had been obtained to characterize the gas stream and Orsat analysis provided O2 and CO2 concentrations for the test periods when the CEMs were not operating. 3.3.1 FTIR Results Gas phase and sample concentration data were analyzed for the presence of HAPs and other species. All spectra were visually inspected and absorbance bands were identified. Then spectra were analyzed, using procedures developed by Entropy, to determine concentrations of any species detected. These results are presented in Tables 3-2 and 3-3. Maximum possible (minimum detectible) concentrations were calculated for undetected HAPs. These results are presented in Tables 3-4, 3-5 and 3-6. 3.3.1.1 Gas Phase Results -- Analysis of the gas phase spectra revealed the samples were composed primarily of:
water vapor
CO2
SO2 was detected at concentrations between 155 and 250 ppm at the
air preheater outlet and at concentrations between 155 and 206 ppm
at the baghouse outlet.
NO was detected at concentrations between 30 and 49 ppm.
NO2 and N2O were both detected but not quantified because Entropy
does not currently have quantitative reference spectra for these
species.
CO was detected but not quantified using FTIR. CEM data indicate
the CO concentration was between 11 and 22 ppm.
Even though water interference was strong in the gas phase samples and the condenser removed NH3, CH2O, and HCl, these species, which were detected in Tenax® samples, would have been detected in the hot/wet samples if present in the flue gas at concentrations at one ppm or higher. Table 3-2 presents calculated concentrations for SO2 and NO. The errors for NO in the hot/wet spectra are large because of interference from water. The concentrations for NO calculated from the condenser and PermaPure® spectra are more accurate, but interference from water was still significant. The concentrations shown in Table 3-2 represent averages of results from up to four spectra. The average results represent concentrations during the times given in Table 3-1 when gas phase samples were collected. A set of subtracted spectra was generated so that maximum possible (minimum detectible) concentrations could be calculated for HAPs that were not identified in the sample stream. Reference spectra of water vapor, SO2, NO and CO2 were scaled and subtracted from each of the field spectra. The remaining base lines were then analyzed for every compound represented in the quantitative spectral library to determine the maximum possible concentrations of HAPs that went undetected. The calculations were performed according to the procedures described in Section 4.6.3. For any undetected compound to be present in the sample there is an upper limit to its possible concentration. This is the quantity that is presented in Tables 3-4 to 3-6 as the maximum possible concentration. For a HAP to have been present in the gas phase samples, its concentration must have been below the calculated maximum possible concentration. Results for hot/wet and dry (treated with the condenser or PermaPure® dryers) spectra are presented in Tables 3-4 and 3-5 respectively. The results are averages of the calculated values for all of the spectra over the 3 sample runs. The hot/wet gas phase spectra are the most difficult to analyze because of strong interference from water vapor. Even so, in results from the hot/wet gas phase data, 91 compounds gave minimum detectible concentrations below 10 ppm, and of these, 72 are below 5 ppm, and 28 are 1 ppm or lower. Previously, Entropy developed program files to analyze for HAPs in FTIR spectra of samples extracted from a coal-fired boiler stack. Statistical analysis showed that the programs were successful in measuring some HAPs in hot/wet and condenser samples.[1] The same programs were used to analyze the data obtained in this test. The results of this analysis are presented in Appendix C. 3.3.1.2 Sample Concentration Results -- The sample concentration spectra represent integrated samples collected over each four hour run. Water vapor, CO2, CO, SO2, NO, N2O, and the following compounds were detected:
Formaldehyde (CH2O) was detected in the sample from Runs 1 and 3 at
the air preheater outlet.
Methane was detected in the ambient samples from the air preheater
outlet and from Run 1 at the air preheater outlet.
Hydrogen chloride (HCl) was detected in samples from Runs 2 and 3
at the air preheater outlet, an ambient sample from the air
preheater outlet, samples from Runs 3 and 4 at the stack, and one
ambient sample at the stack.
Ammonia (NH3), was detected in three of the ambient samples, in
samples from Runs 1 and 2 at the air preheater outlet, and Runs 2
and 3 at the stack.
Toluene was detected in samples from Runs 2, 3 and 4 at the air
preheater outlet and the stack. This assignment is based on the
observance of weak absorbances.
Freon 11 (CCL3F) was detected in the samples from Run 1 at the air
preheater outlet and the stack, and one ambient sample from each
location.
Hydrogen cyanide (HCN) was detected in the samples from all four
runs at the stack. This assignment was based on the observance of
weak absorbances and was complicated by interference with CO2.
Evidence of hexane was observed in samples from both locations and
the ambient samples. Absorbances similar to hexane are often
observed in spectra of desorbed samples. These features may be due
to a mixture of alkane hydrocarbons, including hexane, the sum of
whose spectra gives absorbances which appear similar to hexane.
A cyclic siloxane compound was detected that Entropy first
identified in spectra of samples taken at the coal-fired boiler
validation test.1 This compound was shown to be a product of a
reaction between HCl or water vapor in the gas stream and materials
in the filter housing of the Method 5 box. Entropy took steps to
eliminate this source of contamination and the siloxane, if it is
a contaminant is present at very low levels relative to the
validation data.
An absorbance band was observed that could tentatively be assigned
to either Chlorobenzene or o-xylene. The identification is
complicated by interference from CO2.
Table 3-3 presents calculated concentrations of formaldehyde, methane, ammonia, HCl, HCN and hexane from the sample concentration test runs. In- stack concentrations are also given for the same species. In-stack concentrations were determined by dividing the in-cell concentration by the concentration factor (Section 4.6.4) and do not take into account adsorption/ desorption efficiencies of these volatile species. The values in Table 3-3 represent lower limits on the concentrations for these species. Upper limits are provided by the gas phase data (Tables 3-4 and 3-5). Other absorbance bands were also observed which remain unidentified. None of these bands were attributed to HAPs for which Entropy currently has reference spectra. When these bands are identified, it should become clear whether they are due to emissions from the process or were formed by conditions unrelated to the process (i.e. by contamination). These bands do not consistently appear in every sample. Table 3-6 presents maximum possible concentrations calculated for sample concentration spectra. Analysis programs for analyzing Tenax® samples were developed for the validation test at the coal-fired boiler. These programs were used to analyze the data from TNP and the results are presented in Appendix C. Results are presented only for those compounds that Entropy demonstrated can be measured with thermal desorption FTIR using Tenax® . 3.3.2 Instrumental and Manual Test Results Table 3-7 presents the results of the CEM and manual tests conducted as described in Section 4.3. Table 3-8 summarizes the results presented in Table 3-7. All CEM concentration results presented in Table 3-8 were determined from the average gas concentration measured during the run and adjusted based on the pre- and post-run calibration results (Equation 6C-1 presented in EPA Method 6C, Section 8). Although not required by Methods 10 and 25A, the same data reduction procedures as those in Method 3A were used for the CO and HC determinations to ensure the data quality. All measurement system calibration bias and calibration drift checks for each test run met the applicable specifications contained in the test methods. Both Method 3 and 3A O2 and CO2 results are presented for the inlet location. The Method 3 results for the baghouse inlet and stack locations were used in all flow rate computations. The CO and HC emission rates were computed using the averaged concentration measurements over the course of each test run period, the flue gas volumetric flow rate, and the appropriate conversion factors. Because Method 25A requires that the measurement be made on a wet basis, the wet flow rate results in units of "wet standard cubic feet per minute, wscfm" were used to compute HC emission rates (lb/hr as methane). The CO emission rates were computed using the dry basis concentration and flow rate data. Comparison of the HC measurements indicates a negligible difference in the levels across the baghouse. 3.3.3 Process Results Table 3-9 shows the coal analysis results for two days the plant was tested; Table 3-10 shows the trace element analysis of the Walnut Creek lignite. Figures 3-1 through 3-9 present the process data in graphical form. The raw data used to generate these figures are presented in Appendix B. Figures 3-1, 3-4, and 3-7 present the fuel flow and Unit Two performance for Runs 1, 2, and 3, respectively. Figures 3-2, 3-5, and 3-8 present the stack continuous emission monitor results for Runs 1, 2, and 3, respectively. Figures 3-3, 3-6, and 3-9 present the baghouse performance results. The following occurrences in the process were noted by the observer as potentially affecting the test results:
(1) Before the test, plant personnel installed a continuous strip-chart
reading device on the stack to record the stack gas exit
temperature. The thermocouple for this measurement had previously
been used for a constant (non-recorded) readout rather than a
continuous readout. Test personnel on the stack checked the unit's
operation before each day of testing. After the test was
completed, Tnp personnel removed the recording chart and delivered
it to RTI. Upon examining the chart it was found that the
instrument had stopped recording at 6:00 a.m. on 5/13/93 (between
Runs 2 and 3). Although the data from the recorder did not follow
the trend of the baghouse inlet temperature, the averages of the
two data points differed from each other by only a few degrees
Fahrenheit (this difference was confirmed by telephone with the
Entropy test team who were also monitoring the stack gas exit
temperature). This situation led to suspicion concerning the
accuracy of the baghouse exit temperature readings. The data
suggest two possible explanations:
The thermocouple measuring the baghouse exit temperature was
placed unsuitably in the duct and not reading a true
temperature; or
The thermocouple was mis-identified, for example, a chromel-
alumel thermocouple was mistaken for a copper-constantan
thermocouple.
(2) During all three Runs, the average differential pressure across the
fabric filter was greater than 5 in. w.g. This high-pressure drop
resulted in continuous cleaning cycles. Plant operators said that
this occurrence is normal after a planned outage (which occurred
from 3/19/93 to 4/11/93). Based on past experience, the operators
said that the differential pressure will eventually drop to about
3.8 in. w.g., resulting in approximately 10 cleaning cycles per 24
hours rather than about 60 cleaning cycles per 24 hours, which
occurred during the test.
(3) During Run 2 (9:20 a.m. to 1:20 p.m., 5/13/93), a soot blow
occurred. Soot blowing involves using steam to remove soot from
the tubes in the convective backpass and air preheater sections.
The soot blow occurred between 9:00 and 11:45 a.m. The soot blow
had the following effects on the data.
Preheater inlet temperature - Dropped because of better
thermal efficiency (greater heat transfer) due to less soot on
boiler tubes
Baghouse inlet and outlet temperature - Even though the
baghouse outlet temperature was not accurate, it did follow
the same trend as the baghouse inlet temperature which
increased. This increase was caused by hot soot being pulled
through the baghouse.
Opacity - The increase in opacity was expected because of the
extra particulate loading from the soot blow.
(4) During Run 3 (2:00 p.m. to 6:00 p.m., 5/13/93), another soot blow
occurred, between 4:11 and 6:00 p.m. (see potential effects above).
(5) Upon analyzing the baghouse individual compartment differential
pressures, an unusual pattern was observed. The compartments show
increases in differential pressures as the flue gas flows from
baghouse inlet to outlet. From an overhead view of the baghouse
the compartments are arranged in a rectangular pattern
(Figure 2-2). When asked about this
pattern, the plant superintendent stated it resulted from the
physical configuration of the flue gas duct work going to each
compartment. Similar patterns have been observed at other
baghouses when dust distribution among the compartments was uneven.
4.0 SAMPLING AND ANALYTICAL PROCEDURESThe FTIR analysis is done using two different experimental techniques. The first, referred to as direct gas phase analysis, involves transporting the gas stream to the sample manifold so it can be sent directly to the infrared cell. This technique provides a sample similar in composition to the flue gas stream at the sample point location. Some compounds may be affected because of contact with the sampling system components or reactions with other species in the gas. A second technique, referred to as sample concentration, involves concentrating the sample by passing a measured volume through an absorbing material (Tenax®) packed into a U-shaped stainless steel collection tube. After sampling, the tube is heated to desorb any collected compounds into the FTIR cell. The desorbed sample is then diluted with nitrogen to one atmosphere total pressure. Concentrations of any species detected in the absorption cell are related to flue gas concentrations by comparing the volume of gas collected to the volume of the FTIR cell. Desorption into the smaller FTIR cell volume provides a volumetric concentration related to the volume sampled. This, in turn, provides a corresponding increase in sensitivity for the detection of species that can be measured using Tenax®. Sample concentration makes it possible to achieve lower detection limits for some HAPs. Infrared absorbance spectra of gas phase and concentrated samples were recorded and analyzed. In conjunction with the FTIR sample analysis, measurements of HC, CO, O2, and CO2 were obtained using gas analyzers. Components of the emission test systems used by Entropy for this testing program are described below. 4.1 EXTRACTIVE SYSTEM FOR DIRECT GAS PHASE ANALYSIS Two extractive systems (Figure 4-1) were used to transport the gas stream directly to the FTIR spectrometer; One at the air preheater outlet (baghouse inlet) and the other at the baghouse outlet. Gas samples from each sampling system were sequentially introduced to the absorption cell for analysis. 4.1.1 Sampling System Flue gas was extracted through a stainless steel probe. A Balston® particulate filter rated at 1 micron was installed at the inlet of the sample probe. Heated 3/8 in O.D. Teflon® sample line connected the probe to the heated sample pump (KNF Neuberger, Inc. model number N010 ST.111) located inside the mobile laboratory. One hundred fifty ft of sample line were sufficient to reach the air preheater outlet location while 300 feet of line were required to reach the port located at the baghouse outlet. The temperature of the sampling system components was maintained at about 300 degF. Digital temperature controllers were used to control and monitor the temperature of the transport lines. All connections were wrapped with electric heat tape and insulated to ensure that there were no "cold spots" in the sampling system where condensation might occur. All components of the sample system were constructed of Type 316 stainless steel or Teflon® . The heated sample flow manifold, located in the FTIR truck, included a secondary particulate filter and valves that allowed the operator to send sample gas directly to the absorption cell or through a gas conditioning system. The extractive system can deliver three types of samples to the absorption cell. Sample sent directly to the FTIR cell is considered unconditioned, or "hot/wet." This sample is thought to be most representative of the actual effluent composition. The removal of water vapor from the gas stream before analysis was sometimes desirable; therefore, a second type of sample was provided by directing gas through a condenser system. The condenser employed a standard Peltier dryer to cool the gas stream to approximately 38 degF. The resulting condensate was collected in two traps and removed from the conditioning system with peristaltic pumps. This technique is known to leave the concentrations of inorganic and highly volatile compounds very near to the (dry-basis) stack concentrations. A third type of sample was obtained using a series of PermaPure® dryers. This system utilized a network of semi-permeable membranes. Water vapor was drawn through the membrane walls by a concentration gradient, which was established by a counter flow of dry air along the outside of the membrane walls. In addition to protecting the absorption cell, water removal relieved spectral interferences, which could limit the effectiveness of the FTIR analysis for particular compounds. 4.1.2 Analytical System The FTIR equipment used in this test consists of a medium-resolution interferometer, heated infrared absorption cell, liquid nitrogen cooled mercury cadmium telluride (MCT) broad band infrared detector, and computer (Figure 4-2). The interferometer, detector, and computer were purchased from KVB/Analect, Inc., and comprise their base Model RFX-40 system. The nominal spectral resolution of the system is one wavenumber (1 cm-1). Samples were contained in a model 5-22H infrared absorption cell manufactured by Infrared Analysis, Inc. The inside walls and mirror housing of the cell were Teflon® coated. Cell temperature was maintained at 240 degF using heated jackets and temperature controllers. The absorption path length of the cell was set at 22 meters. 4.1.3 Sample Collection Procedure During all four test runs, direct gas phase analysis was performed concurrently with the sample concentration runs. One gas phase analysis run was performed at each of two locations; the inlet and outlet of the baghouse. Gas phase analysis was not performed at the stack. During each run, flue gas continuously flowed through the heated system to the sample manifold in the FTIR truck. A portion of the gas stream was diverted to a secondary manifold located near the inlet of the FTIR absorption cell (Figure 4-2). The cell was filled with sample to ambient pressure and the FTIR spectrum recorded. After analysis, the cell was evacuated so that a subsequent sample could be introduced. The process of collecting and analyzing a sample, then evacuating the cell to prepare for the next sample required less than 10 minutes. During each run, about 12 gas phase samples were analyzed at each location. Sample concentration was performed using the adsorbent material Tenax®, followed by thermal desorption into the FTIR cell. The sample collection system employed equipment similar to that of the Modified Method 5 sample train. 4.2.1 Sampling System Figure 4-3 depicts the apparatus used in this test program. Components of the sampling train included a heated stainless steel probe, heated filter and glass casing, stainless steel air-cooled condenser, stainless steel adsorbent trap in an ice bath, followed by two water-filled impingers, one knockout impinger, an impinger filled with silica gel, a sample pump, and a dry gas meter. All heated components were kept at a temperature above 120 degC to ensure no condensation of water vapor within the system. The stainless steel condenser coil was used to pre-cool the sample gas before it entered the adsorbent trap. The trap was a specially designed stainless steel U- shaped collection tube filled with 10 g of Tenax® and plugged at both ends with glass wool. Stainless steel was used for the construction of the adsorbent tubes because it gives a more uniform and more efficient heat transfer than glass. Each sampling run was 4-hours at approximately 0.12 to 0.17 dcfm for a total sampled volume of about 30 to 40 dcf. The sampling rate depended on the sampling train used and was close to the maximum that could be achieved. Collection times provided a volumetric concentration that was proportional to the total volume sampled. The resulting increase in sensitivity allowed detection to concentrations below 1 ppm for some HAPs. 4.2.2 Analytical System Before analysis condensed water vapor was removed from the collection tubes using a dry nitrogen purge for about 15 minutes. Sample analyses were performed using thermal desorption-FTIR. The sample tubes were wrapped with heat tape and placed in an insulated chamber. One end of the tube was connected to the inlet of the evacuated FTIR absorption cell. The same end of the tube that served as the inlet during the sample run served as the outlet for the thermal desorption. Gas samples were desorbed by heating the Tenax® to 250 degC. A preheated stream of UPC grade nitrogen was passed through the adsorbent and into the FTIR absorption cell. About 7 liters of nitrogen (at 240 degF) carried the desorbed gases to the cell and brought the total pressure of the FTIR sample to ambient pressure. The infrared absorption spectrum was then recorded. The purging process was repeated until no evidence of additional sample desorption was noted in the infrared spectrum. 4.2.3 Sample Collection Procedure During each 4-hour run, concentrated samples were collected simultaneously at the baghouse inlet and the stack. A sample concentration apparatus was set up at each location and ambient samples (Section 5.4.1) were collected to check for contamination in each train. Entropy performed leak checks of the system and the start time of each run was synchronized at both locations. Sample flow, temperature of the heated box, and the tube outlet temperature were monitored continuously and recorded at 10-minute intervals. At the end of each run, sampling was interrupted and the collection tube was removed. The open ends were tightly capped and the tube was stored on ice until it was analyzed. The tubes were analyzed within 12 hours after the end of the sample run. 4.3 CONTINUOUS EMISSIONS MONITORING Entropy's extractive measurement system and the sampling and analytical procedures used for the determinations of hydrocarbons (HC), carbon monoxide (CO), oxygen (O2), and carbon dioxide (CO2) conform with the requirements of EPA Test Methods 25A, 10, and 3A, respectively, of 40 CFR 60, Appendix B. Two heated extractive sampling systems and a single set of gas analyzers were used to analyze flue gas samples extracted at the baghouse inlet and outlet sample point locations. The analyzers received gas samples delivered sequentially from the two sample point locations. These gas analyzers require that the flue gas be conditioned to eliminate any possible interference (i.e., water vapor and particulate matter) before being transported and analyzed. All components of the sampling system that contact the gas sample are Type 316 stainless steel and Teflon®. A gas flow distribution manifold downstream of the heated sample pump was used to control the flow of sample gas to each analyzer. A refrigerated condenser removed water vapor from the sample gas analyzed by the CO, CO2, and O2 analyzers. Wet sample was supplied to the HC analyzer (Method 25A requires a wet basis analysis). The condenser is operated at approximately 38 degF. Condensate is continuously removed from the traps in order to minimize contact with the gas sample. The sampling system includes a calibration gas injection point immediately upstream of the analyzers for the calibration error checks and also at the outlet of the probe for the sampling system bias and calibration drift checks. The mid- and high-range calibration gases are certified by the vendor according to EPA Protocol 1 specifications. Methane in air was used to calibrate the HC analyzer. A computer-based data acquisition system is used to provide an instantaneous display of the analyzer responses, as well as compile the measurement data collected each second, calculate data averages over selected time periods, calculate emission rates, and document the measurement system calibrations. Table 4-1 presents a list of the analyzers that Entropy used during the test program to quantify the gas concentration levels at the sample point locations. Because the sampling location at the preheater outlet was only five feet upstream of the point where the primary air outlet and the secondary air outlet join, there was a potential for flow disturbances at the sampling point. Velocity traverse measurements were taken through all six ports available on the two ducts using a standard S-type pitot tube and also a 3- dimensional pitot probe. The 3-D probe was utilized to obtain measurements through each port in an effort to identify off-axial flow. Velocity traverse data were obtained at 14 points through each port (42 points in each duct) to establish a flow profile in the sampling region. Once two suitable ports were chosen, flue gas was sampled with the gas phase probe to establish the point of lowest O2 concentration. The sample concentration probe was inserted adjacent to the gas phase probe. A pitot tube was also inserted in the third port to the same depth as the other two probes. The pitot measurements helped to verify that flow conditions were stable during each of the test runs. P measurements were recorded at ten minute intervals. Sample flow through the dry gas meter, the temperatures of the meter box and the tube outlet were also monitored and recorded every 10 minutes. The 3-D probe had been previously calibrated in wind tunnel experiments. Calibration data that had been obtained at wind velocities of 40 and 50 ft/sec was averaged and combined with the 3-D data to help determine flow velocity at the air preheater outlet. The stack location satisfied EPA Method 1 criteria; therefore, the determinations at the stack were made in accordance with EPA Methods 1, 2, and 3. The wet-bulb/dry-bulb technique was used for the measurement of the flue gas moisture. Pitot traverse point locations and the measurements made at these points are presented on the data sheets included in Appendix A. Flow determinations for Runs 1 and 2 involved conducting velocity traverses both before and after each 4-hour run, and averaging the pre- and post- test results. Because Runs 3 and 4 were performed on the same day, a single velocity traverse was performed before Run 3 and following Run 4. During the test Runs, a representative from Research Triangle Institute (RTI) monitored the process operations so that emissions test data could be correlated with process data. Process observations are described in Section 3.3.3. 4.6.1 Description of K-Matrix Analyses K-type calibration matrices were used to relate absorbance to concentration. Several descriptions of this analytical technique can be found in the literature[2]. The discussion presented here follows that of Haaland, Easterling, and Vopicka[3]. For a set of m absorbance reference spectra of q different compounds over n data points (corresponding to the discrete infrared wavenumber positions chosen as the analytical region) at a fixed absorption pathlength b, Beer's law can be written in matrix form as
The quantity which is sought in the design of this analysis is the matrix K, since if an approximation to this matrix, denoted by K, can be found, the concentrations in a sample spectrum can also be estimated. Using the vector A* to represent the n measured absorbance values of a sample spectrum over the wavenumber region(s) of interest, and the vector C to represent the j estimated concentrations of the compounds comprising the sample, C can be calculated from A* and K from the relation
Here the superscript t represents the transpose of the indicated matrix, and the superscript -1 represents the matrix inverse.
The standard method for obtaining the best estimate K is to minimize the square of the error terms represented by the matrix E. The equation represents the estimate K which minimizes the analysis error. Reference spectra for the K-matrix concentration determinations were de- resolved to 1.0 cm-1 resolution from existing 0.25 cm-1 resolution reference spectra. This was accomplished by truncating and re-apodizing[4] the interfer- ograms of single beam reference spectra and their associated background interferograms. The processed single beam spectra were recombined and converted to absorbance (see Section 4.3). 4.6.2 Preparation of Analysis Programs To provide accurate quantitative results, K-matrix input must include absorbance values from a set of reference spectra which, added together, qualitatively resemble the appearance of the sample spectra. For this reason, all of the Multicomponent analysis files included spectra representing interferant species and criteria pollutants present in the flue gas. Several factors affect the detection and analysis of an analyte in the stack gas matrix. One is the composition of the stack gas. The major spectral interferant in coal-fired boiler effluent are water, SO2 and CO2. At CO2 concentrations of about 10 percent and higher, weak absorbance bands that are normally not visible begin to emerge. Some portions of the FTIR spectrum were not available for analysis because of extreme absorbance from water and CO2, but most compounds exhibit at least one absorbance band that is suitable for analysis. Significant amounts of NO, and NO2 were also present in the samples and these species needed to be accounted for in any analytical program. A second factor affecting analyses is the number of analytes that are to be detected because the program becomes more limited in distinguishing overlapping bands as the number of species in the sample increases. A third factor depends on how well the sample spectra can be modeled. The best analysis can be made when reference spectra are available to account for all of the species detected in the sample. When reference spectra are not available for a compound which has been identified, then it becomes more difficult to quantify other species. A set of Multicomp program files had been previously prepared for analysis of data collected at a coal-fired utility for the purpose of performing statistical validation testing of the FTIR methods. Separate programs were prepared to measure 47 different compounds. Four baseline subtraction points were specified in each analytical region, identifying an upper and a lower baseline averaging range. The absorbance data in each range were averaged, a straight baseline was calculated through the range midpoint using the average absorbance values, and the baseline was subtracted from the data prior to K-matrix analysis. Before K-matrix analysis was applied to data all of the spectra were inspected to determine what species had been detected. Program files were constructed that included reference spectra representing the detected species and were then used to calculate concentrations of the detected species. Sample concentration spectra were also analyzed using program files that were shown by the validation testing to be suitable for measuring some HAPs. 4.6.3 Error Analysis of data The principal constituents of the gas phase samples were water, CO2, NO, and NO2. A program file was prepared to quantify each of these compounds. Other than these species and N2O no major absorbance features were observed in the spectra. After concentrations of the main constituents were determined, the appropriate standard was scaled and subtracted from the spectrum of the sample mixture. This helped verify the calculated values. New spectra were generated from the original absorbance spectra by successively subtracting scaled standard spectra of water, CO2, NO, and NO2. The resulting "subtracted" spectra were analyzed for detectible absorbencies of any HAPs and, for undetected species, the maximum possible concentrations that could have been present in the samples. Maximum possible (minimum detectible) concentrations were determined in several steps. The noise level in the appropriate analytical region was quantified by calculating the root mean square deviation (RMSD) of the baseline in the subtracted spectrum. The RMSD was multiplied by the width (in cm-1) of the analytical region to give an equivalent "noise area" in the subtracted spectrum. This value was compared to the integrated area of the same analytical region in a standard spectrum of the pure compound. The noise was calculated from the equation:
where:
RMSD = Root mean square deviation in the absorbance values within a
region.
n = Number of absorbance values in the region.
Ai = Absorbance value of the ith data point in the analytical
region.
AM = Mean of all the absorbance values in the region.
If a species is detected, then the error in the calculated concentration is given by:
where:
Eppm = Noise related error in the calculated concentration, in ppm.
x2 = Upper limit, in cm-1, of the analytical region.
x1 = Lower limit, in cm-1, of the analytical region.
AreaR = Total band area (corrected for path length, temperature, and
pressure) in analytical region of reference spectrum of
compound of interest.
CONR = Known concentration of compound in the same reference
spectrum.
This ratio provided a concentration equivalent to measured area in the subtracted spectrum. For instances when a compound was not detected, the value Eppm was equivalent to the minimum detectible concentration of that (undetected) species in the sample. Some concentrations given in Tables 3-4 to 3-6 are relatively high (greater than 10 ppm) and there are several possible reasons for this.
The reference spectrum of the compound may show low absorbance at
relatively high concentrations so that its real limit of detection is
high. An example of this may be acetonitrile.
The region of the spectrum used for the analysis may have residual
bands or negative features resulting from the spectral subtraction.
In these cases the absorbance of the reference band may be large at
low concentrations, but the RMSD is also large (see Equation 7).
The chosen analytical region may be too large, unnecessarily including
regions of noise where there is no absorbance from the compound of
interest.
In the second and third cases the stated maximum possible concentration may be lowered by choosing a different analytical region, generating better subtracted spectra, or by narrowing the limits of the analytical region. Entropy has already taken these steps to lower the calculated minimum detectible concentrations. Additional improvement may be gained by regenerating reference spectra of some compounds using longer path lengths. If more improvements can be made, they will be included in the final report. 4.6.4 Concentration Correction Factors Calculated concentrations in sample spectra were corrected for differences in absorption pathlength between the reference and sample spectra according to the following relation:
Corrections for variation in sample pressure were considered, and found to affect the indicated HAP concentrations by no more that one to two percent. Since this is a small effect in comparison to other sources of analytical error, no sample pressure corrections were made. 4.6.5 Analysis of Sample Concentration Spectra Sample concentration spectra were analyzed in the same manner as spectra of the gas phase samples. To derive flue gas concentrations it was necessary to divide the calculated concentrations by the concentration factor (CF). As an illustration, sampling 10 ft3 (about 283 liters) of gas and desorbing into the FTIR cell volume of approximately 8.5 liters gives a concentration factor of about 33. If a compound is detected at a concentration of 50 ppm in the cell, then its corresponding flue gas concentration is about 1.5 ppm. The total volume sampled was determined from the following relation:
The concentration factor, CF, was then determined using Vflue and the volume of the FTIR cell (Vcell) which was measured at an absolute temperature (Tcell) of about 300 K: Finally, the in-stack concentration was determined using CF and the calculated concentration of the sample contained in the FTIR cell, Ccell. 5.0 QUALITY ASSURANCE/QUALITY CONTROL ACTIVITIESQuality assurance (QA) is defined as a system of activities that provides a mechanism of assessing the effectiveness of the quality control procedures. It is a total integrated program for assuring the reliability of monitoring and measurement data. Quality control (QC) is defined as the overall system of activities designed to ensure a quality product or service. This includes routine procedures for obtaining prescribed standards of performance in the monitoring and measurement process. The specific internal QA/QC procedures that were used during this test program to facilitate the production of useful and valid data are described in this section. Each procedure was an integral part of the test program activities. Section 5.1 covers method-specific QC procedures for the manual flue gas sampling. Section 5.2 covers the QC procedures used for the instrumental methods. QC checks of data reduction, validation and reporting procedures are covered in Section 5.3, and corrective actions are discussed in Section 5.4. 5.1 QC PROCEDURES FOR MANUAL FLUE GAS TEST METHODS This section details the QC procedures that were followed during the manual testing activities. 5.1.1 Velocity/Volumetric Flow Rate QC Procedures The QC procedures for velocity/volumetric flow rate determinations followed guidelines set forth by EPA Method 2. Incorporated into this method are sample point determinations by EPA Method 1. Gas moisture determination was approximated using the wet bulb-dry bulb technique. The following QC steps were followed during these tests:
The S-type pitot tube was visually inspected before sampling.
Both legs of the pitot tube were leak checked before and after
sampling.
Proper orientation of the S-type pitot tube was maintained while
making measurements. The roll and pitch axis of the S-type pitot
tube was maintained at 90deg to the flow.
The magnehelic set was leveled and zeroed before each run.
The pitot tube/manometer umbilical lines were inspected before and
after sampling for leaks and moisture condensate (lines were cleared
if found).
Cyclonic or turbulent flow checks were performed prior to testing the
source.
Reported duct dimensions and cross-sectional duct area were verified
by on-site measurements.
If a negative static pressure was present at sampling ports, checks
were made for air in-leakage at the sample port which could have
resulted in possible flow and temperature errors. Leaks were sealed
when found.
The stack gas temperature measuring system was checked by observing
ambient temperatures prior to placement in the stack.
The QC procedures that were followed in regards to accurate sample gas volume determination are:
The dry gas meter is fully calibrated every 6 months using an EPA
approved intermediate standard.
Pre-test and post-test leak checks were completed and were less than
0.02 cfm or 4 percent of the average sample rate.
The gas meter was read to a thousandth (.001) of a cubic foot for the
initial and final readings.
Readings of the dry gas meter, meter orifice pressure ( H), and meter
temperatures were taken every 10 minutes during sample collection.
Accurate barometric pressures were recorded at least once per day.
Post-test dry gas meter checks were completed to verify the accuracy
of the meter full calibration constant (Y).
5.1.2 Sample Concentration Sampling QC Procedures QC procedures that allowed representative collection of organic compounds by the sample concentration sampling system were:
Only properly cleaned glassware and prepared adsorbent tubes that had
been kept closed with stainless steel caps were used for any sampling
train.
The filter, Teflon® transfer line, and adsorbent tube were maintained
at +-10degF of the specified temperatures.
An ambient sample was analyzed for background contamination.
Clean sample tubes were analyzed for contamination prior to their use
in testing.
5.1.3 Manual Sampling Equipment Calibration Procedures 5.1.3.1 Type-S Pitot Tube Calibration -- EPA has specified guidelines concerning the construction and geometry of an acceptable Type-S pitot tube. If the specified design and construction guidelines are met, a pitot tube coefficient of 0.84 is used. Information pertaining to the design and construction of the Type-S pitot tube is presented in detail in Section 3.1.1 of EPA document 600/4-77-027b. Only Type-S pitot tubes meeting the required EPA specifications were used. The pitot tubes were inspected and documented as meeting EPA specifications prior to field sampling. 5.1.3.2 Temperature Measuring Device Calibration -- Accurate temperature measurements are required during source sampling. The bimetallic stem thermometers and thermocouple temperature sensors used during the test program were calibrated using the procedure described in Section 3.4.2 of EPA document 600/4-77-027b. Each temperature sensor is calibrated at a minimum of three points over the anticipated range of use against a NIST-traceable mercury-in-glass thermometer. All sensors were calibrated prior to field sampling. 5.1.3.3 Dry Gas Meter Calibration -- Dry gas meters (DGMs) were used in the sample trains to monitor the sampling rate and to measure the sample volume. All DGMs were fully calibrated to determine the volume correction factor prior to their use in the field. Post-test calibration checks were performed as soon as possible after the equipment was returned as a QA check on the calibration coefficients. Pre- and post-test calibrations should agree within 5 percent. The calibration procedure is documented in Section 3.3.2 of EPA document 600/4-77-237b. 5.1.3.4 3-Dimensional Probe Calibration -- The following QC procedures were performed when using the 3-dimensional probe.
The barometric pressure was recorded daily.
The entire sampling system was leak checked prior to each run.
The direction of gas flow will be determined before sampling.
The angle finder was determined to be working properly.
The manometers were leveled and zeroed every day.
The probe was positioned at the measurement point and rotated in the
gas stream until zero deflection is indicated for the yaw angle;
this null position occurs when P2 = P3. Each yaw angle reading from
the protractor or other angle measuring device was recorded.
Holding the null reading position, readings were taken and recorded
for the (P1 - P2) and (P4 P5). Record the duct pressure or the (P2
- Pbar).
Repeat the procedure at each of the measurement points.
The pitch angle was determined from the F1 calibration curve and F2
was determined from the F2 calibration curve.
Calibration curves were generated from the procedures outlined in
Draft Method 2E.
5.2 QC PROCEDURES FOR INSTRUMENTAL METHODS The flue gas was analyzed for carbon monoxide (CO), oxygen (O2), carbon dioxide (CO2) and total hydrocarbons (THC). Prior to sampling each day a pre- test leak check of the sampling system from the probe tip to the heated manifold was performed and was less than 4 percent of the average sample rate. Internal QA/QC checks for the CEM systems are presented below. 5.2.1 Daily Calibrations, Drift Checks, and System Bias Checks Method 3A require that the tester : (1) select appropriate apparatus meeting the applicable equipment specifications of the method, (2) conduct an interference response test prior to the testing program, and (3) conduct calibration error (linearity), calibration drift, and sampling system bias determinations during the testing program to demonstrate conformance with the measurement system performance specifications. The performance specifi- cations are identified in the following table. -----------------------------------------------------------------------------
Table 5-1. Instrumental Test Method Specifications
PERFORMANCE TEST SPECIFICATION
---------------- -------------
Analyzer Calibration Error +- 2% of span for zero, mid-, and
high-range calibration gases
Sampling System Bias +- 5% of span for zero and upscale
calibration gases
Zero Drift +- 3% of span over test run period
Upscale Calibration Drift +- 3% of span over test run period
Interference Check +- 7% of the modified Method 6 result
for each run
-----------------------------------------------------------------------------
A three-point (i.e., zero, mid-, and high-range) analyzer calibration error check is conducted before initiating the testing by injecting the calibration gases directly into the gas analyzer and recording the responses. Zero and upscale calibration checks are conducted both before and after each test run in order to quantify measurement system calibration drift and sampling system bias. Upscale is either the mid- or high-range gas, whichever most closely approximates the flue gas level. During these checks, the calibration gases are introduced into the sampling system at the probe outlet so that the calibration gases are analyzed in the same manner as the flue gas samples. Drift is the difference between the pre- and post-test run calibration check responses. Sampling system bias is the difference between the test run calibration check responses (system calibration) and the initial calibration error responses (direct analyzer calibration) to the zero and upscale calibration gases. If an acceptable post-test bias check result is obtained but the zero or upscale drift result exceeds the drift limit, the test run result is valid; however, the analyzer calibration error and bias check procedures must be repeated before conducting the next test run. A run is considered invalid and must be repeated if the post-test zero or upscale calibration check result exceeds the bias specification. The calibration error and bias checks must be repeated and acceptable results obtained before testing can resume. Although not required by Methods 10 and 25A, the same calibration and data reduction procedures required by Method 3A were used for the CO and HC determinations to ensure the quality of reference data. 5.3 QA/QC CHECKS FOR DATA REDUCTION, VALIDATION, AND REPORTING Data quality audits were conducted using data quality indicators which require the detailed review of: (1) the recording and transfer of raw data; (2) data calculations; (3) the documentation of procedures; and (4) the selection of appropriate data quality indicators. All data and/or calculations for flow rates, moisture content, and sampling rates were spot checked for accuracy and completeness. In general, all measurement data have been validated based on the following criteria:
Acceptable sample collection procedures.
Adherence to prescribed QC procedures.
Any suspect data have been identified with respect to the nature of the problem and potential effect on the data quality. Upon completion of testing, the field coordinator was responsible for preparation of a data summary including calculation results and raw data sheets. 5.3.1 Sample Concentration The sample concentration custody procedures for this test program were based on EPA recommended procedures. Because collected samples were analyzed on-site, the custody procedures emphasized careful documentation of sample collection and field analytical data. Use of chain-of-custody documentation was not necessary. Instead, careful attention was paid to the sample identification coding. These procedures are discussed in more detail below. Each spectrum of a sample concentration sample was assigned a unique alphanumeric identification code. For example, Tinl102A designates a desorption spectrum of a Tenax® sample taken at the baghouse inlet (air preheater outlet) during run number one using tube number 02. The A means this was the spectrum of the first desorption from this tube. Every collection tube was inscribed with a tube identification number. The project manager was responsible for ensuring that proper custody and documentation procedures were followed for the field sampling, sample recovery, and for reviewing the sample inventory after each run to ensure complete and up-to-date entries. A sample inventory was maintained to provide an overview of all sample collection activities. Every sample tube was cleaned and checked for contamination before use. The contamination check consisted of desorbing the clean tube and recording its FTIR spectrum. Each of the sample trains was tested for contamination before any test runs were done and after all runs were completed. The trains were set up according to the procedures of Section 4.2, except that the probe was not inserted into the port. Ambient air was drawn through the entire sample concentration apparatus for one hour. This time was sufficient to reveal significant contamination on the sampling system components. The volume of air drawn for the ambient checks was about 10 ft3. The charged ambient tube was stored and analyzed in the same manner as those obtained during testing. If relatively minor contamination was identified from the ambient sample, it was accounted for in the subsequent analyses by using spectral subtraction. Evidence of major contamination was not identified in any instance. Sample flow at the dry gas meter was recorded at 10 minute intervals. Results from the analyzers and the spectra of the gas phase samples provided a check on the consistency of the effluent composition during the sampling period. 5.3.2 Gas Phase Analysis During test Runs 1 and 2 a total of 12 gas phase samples were collected from each location and analyzed. During Run 3, 18 gas phase samples were analyzed from both locations. Each spectrum was assigned a unique file name and a separate data sheet identifying sample location and sampling conditions. A comparison of all spectra in this data set provided information on the consistency of effluent composition and a real-time check on the performance of the sampling system. Effluent was directed through all sampling lines for at least 5 minutes and the CEM's provided consistent readings over the same period before sampling was attempted. This requirement was satisfied any time there was a switch to a different conditioning system or a switch between testing locations. The FTIR was continuously scanning, when the cell was evacuating, to provide a spectral profile of the empty cell. A new sample was not introduced until there was no residual absorbance remaining from the previous one. The FTIR was also continuously scanning during sample collection to provide a real-time check on possible contamination in the system. 5.3.3 FTIR Spectra For a detailed description of QA/QC procedures relating to data collection and analysis, refer to the draft version of the "Protocol For Applying FTIR Spectrometry in Emission Testing" included in Appendix D. A spectrum of the calibration transfer standard (CTS) was recorded at the beginning and end of each data collection session and a leak check of the FTIR cell was performed at the same times. The CTS gas was 100 ppm ethylene in nitrogen. The CTS spectrum provide a check on the operating conditions of the FTIR instrumentation, e.g. spectral resolution and cell path length. Ambient pressure was recorded whenever a CTS spectrum was collected. Two copies of all interferograms and processed spectra of backgrounds, samples, and the CTS were stored on separate computer disks. Additional copies of sample and CTS absorbance spectra were also stored for use in the data analysis. Sample spectra can be regenerated from the raw interferograms, if necessary. FTIR spectra are available for inspection or re-analysis at any future date. Pure, dry ("zero") air was periodically introduced through the sampling system in order to check for contamination or if condensation formed. Once, when water condensed in the manifold, the lines and cell were purged with dry N2, until the sampling could continue. As successive spectra were collected the position and slope of the spectral base line were monitored. If the base line within a data set for a particular sample run began to deviate by more than 5 percent from 100 percent transmittance, a new background was collected. During the course of the testing program, it was the responsibility of the field coordinator and the sampling team members to ensure that all data measurement procedures were followed as specified and that data met the prescribed acceptance criteria. Specific procedures for corrective actions are described above. The only significant problem arose when the first desorption samples were excessively wet. The resulting spectra were adequate but not of good quality. An additional sampling run was added to the schedule and this was the only corrective action taken. 6.0 CONCLUSIONSEntropy conducted an emission test at TNP One Unit Two in Hammond, Texas. Direct gas phase analysis and sample concentration test runs were performed over three days. Gas analyzers were used to measure CO, O2, CO2, and hydrocarbons in the gas streams. Samples were extracted sequentially from the air preheater outlet and the baghouse outlet using the extractive system for gas phase analysis. The sample concentration system was used to collect samples simultaneously at the air preheater outlet and the stack. The gas phase system detected water vapor, CO2, CO, SO2, NO, and NO2. Sample concentration detected formaldehyde, ammonia, HCl, SO2, HCN, methane, hexane, and a cyclic siloxane compound. Ammonia was also detected in the ambient samples collected at the stack. Some absorbance bands remain unidentified. A primary goal of this project was to use FTIR instrumention in a major test program to measure as many HAPs as possible or to place upper limits on their concentrations. Four other electric utilities were tested along with TNP. Utilities present a difficult testing challenge for FTIR techniques because: (1) they are combustion sources so the flue gas contains high levels of moisture and CO2 (both are spectral interferants) and (2) the large volumetric flow rates typical at these facilities lead to mass emissions above regulated limits even for HAPs at very low concentrations. This places great demand on the measurement method to achieve low detection limits. This represents the first attempt to use FTIR spectroscopy in such an ambitious test program. The program made significant achievements and demonstrated some important and fundamental advantages of FTIR spectroscopy as an emissions test method:
Quantitative data were provided for a large number of compounds
using one method.
Software was written to provide concentration and detection limit
results in a timely manner. The same or similar software can be
used for subsequent tests with very little investment of time for
minor modifications or improvements.
Preliminary data (qualitative and quantitative) is provided on-site
in real time.
With little effort at optimization (see below), detection limits in
the ppb range were calculated for 28 HAPs and below 5 ppm for a
total of 72 HAPs using direct gas phase measurements. Sample
concentration provided even lower detection limits for some HAPs.
A positive identification of a compound is unambiguous.
It is appropriate to include some discussion about the "maximum possible concentrations" presented in Tables 3-4 to 3-6. These numbers were specifically not labeled as detection limits because use of that term could be misinterpreted. In FTIR analysis maximum concentrations of non-detects are calculated from the spectra (see Section 4.6.3 and the "FTIR Protocol"). These calculated numbers do not represent fundamental measurement limits, but they depend on a number of factors. For example:
Some instrumental factors
Spectral resolution.
Source intensity.
Detector response and sensitivity.
Path length that the infrared beam travels through the sample.
Scan time.
Efficiency of infrared transmission (through-put).
Signal gain.
Some sampling factors
Physical and chemical properties of compound.
Flue gas composition.
Flue gas temperature.
Flue gas moisture content.
Length of sample line (distance from location).
Temperature of sampling components.
Sample flow.
Instrumental factors are adjustable. For this program instrument settings were chosen to duplicate conditions that were successfully used in previous screening tests and the validation test. These conditions provide speed of analysis, durability of instrumentation, and the best chance to measure a large number of compounds with acceptable sensitivity. Sampling factors present similar challenges to any test method. An additional consideration is that the numbers presented in Tables 3-4 to 3-6 are all higher than the true detection limits that can be calculated from the 1 cm-1 data collected at TNP. This results from the method of analysis: the noise calculations were made only after all spectral subtractions were completed. Each spectral subtraction adds noise to the resulting subtracted spectrum. For most compounds it is necessary to perform only some (or none) of the spectral subtractions before the detection limit can be calculated. With even more sophisticated software it will be possible to automate the process of performing selective spectral subtractions and optimizing the detection limit calculation for each compound of interest. (Such an undertaking was beyond the scope of the current project.) Furthermore, the maximum concentrations are averages compiled from the results of all the spectra collected at given location. A more realistic detection limit is provided by the single spectrum whose analysis gives the lowest calculated value. It would be more accurate to think of maximum concentrations as placing upper boundaries on the HAP detection limits provided by these data. Perhaps the most important sampling consideration is the flue gas composition. In Table 3-4 the maximum concentration is quoted as 6.53 ppm. This was calculated by measuring the noise in the analytical region between 3020 and 3125 cm-1, where benzene has an absorbance band. Benzene exhibits a much stronger infrared band at 673 cm-1, but this band was not used for the analysis because absorbance from CO2 strongly interfered in this analytical region. Using identical sampling components and FTIR instrumental settings at a lower CO2 emission source would provide a detection limit below 1 ppm for benzene for direct gas analysis (even ignoring the consideration discussed in the previous paragraph). There may be some question as to why certain species were not detected, particularly HCl. Table 3-9 presents the coal sampling analysis data (supplied to RTI by TNP) for two days when Entropy sampled. According to the analyses, the coal contained between .02 and .04 percent chlorine and between 1.5 to 1.75 percent sulfur (dry basis). The limestone used in the fluidized bed combustor to control SO2 emissions could also lower chloride emissions by forming CaCl2. The combustion temperature of 1560 degF is above the melting points of CaCl2 and CaSO4, but below the boiling points of both compounds. Even though CaCl2 is water soluble, it should have been a suspended solid at the flue gas temperatures of about 350 degF and removed in the baghouse. Hydrates of CaCl2 should not form at 350 degF. The HCl concentration may actually have been below 750 ppb at the baghouse outlet as given in Table 3-4. The fluidized bed combustion system was effective at lowering the SO2 concentration to about 200 ppm. If the limestone injection was similarly effective at lowering the HCl emissions, then the HCl concentration would probably have been decreased below 1 ppm. If HCl was present above 1 ppm, which is detectible by direct FTIR gas analysis in the wet stream, its measurement could have been affected by the relatively high moisture content (between 16 and 19 percent) of the flue gas. The sampling system configuration (including temperature of components) was chosen because the same configuration was used successfully at other tests. HCl was measured at utilities and other emission sources during the FTIR development project, and it was assumed that flue gas conditions at TNP would be similar. In fact, the flue gas at TNP contained 16 to 19 percent moisture which is significantly higher that the 7 percent moisture encountered at the screening and validation utility tests. High moisture makes HCl measurements more difficult because HCl is water soluble. (Entropy has detected HCl in high moisture streams but only after using dilution.) A higher sampling temperature would not have helped because the sample lines were maintained at 350 degF which was near the gas temperature. The FTIR cell temperature was 250 degF (to keep spectral conditions near those of the reference spectra), but should not have interfered with HCl measurements because no condensation formed in the cell. Previous studies on HCl sampling and measurement in wet streams indicate that high sample flow rates are required to deliver HCl to the measurement system. Entropy has participated in EPRI (Electric Power Research Institute) studies performing FTIR CEM measurements at utilities. In these studies HCl was measured using a sample flow of 10-15 lpm and heated lines at 300 degF. At the TNP test it was not possible to achieve a sample flow rate above 6-7 lpm because Entropy was also delivering sample to gas analyzers. In later FTIR field tests Entropy has performed QA spiking with HCl (and other compounds) to verify sampling system integrity, and this would be a good procedure to include in the test method. The important point to emphasize is that 19 percent moisture presents a sampling system difficulty that any test method must address, not an analytical difficulty. 7.0 REFERENCES
1) "FTIR Method Validation at a Coal-Fired Boiler," EPA Contract No.
68D20163, Work Assignment 2, July, 1993.
2) "Computer-Assisted Quantitative Infrared Spectroscopy," Gregory L.
McClure (ed.), ASTM Special Publication 934 (ASTM), 1987.
3) "Multivariate Least-Squares Methods Applied to the Quantitative Spectral
Analysis of Multicomponent Mixtures," Applied Spectroscopy, 39(10), 73-
84,1985.
4) "Fourier Transform Infrared Spectrometry," Peter R. Griffiths and James
de Haseth, Chemical Analysis, 83, 16-25,(1986), P. J. Elving, J. D.
Winefordner and I. M. Kolthoff (ed.), John Wiley and Sons,.
|


 where:
Ccorr = The pathlength corrected concentration.
Ccalc = The initial calculated concentration (output of the Multicomp
program designed for the compound)
Lr = The pathlength associated with the reference spectra.
Ls = The pathlength (22m) associated with the sample spectra.
Ts = The absolute temperature of the sample gas (388 K).
Tr = The absolute gas temperature at which reference spectra were
recorded (300 to 373 K).
where:
Ccorr = The pathlength corrected concentration.
Ccalc = The initial calculated concentration (output of the Multicomp
program designed for the compound)
Lr = The pathlength associated with the reference spectra.
Ls = The pathlength (22m) associated with the sample spectra.
Ts = The absolute temperature of the sample gas (388 K).
Tr = The absolute gas temperature at which reference spectra were
recorded (300 to 373 K).
 where:
Vflue = Total volume of flue gas sampled.
Vcol = Volume of gas sampled as measured at the dry gas meter after
it passed through the collection tube.
Tflue = Absolute temperature of the flue gas at the sampling location.
Tcol = Absolute temperature of the sample gas at the dry gas meter.
W = Fraction (by volume) of flue gas stream that was water vapor.
where:
Vflue = Total volume of flue gas sampled.
Vcol = Volume of gas sampled as measured at the dry gas meter after
it passed through the collection tube.
Tflue = Absolute temperature of the flue gas at the sampling location.
Tcol = Absolute temperature of the sample gas at the dry gas meter.
W = Fraction (by volume) of flue gas stream that was water vapor.


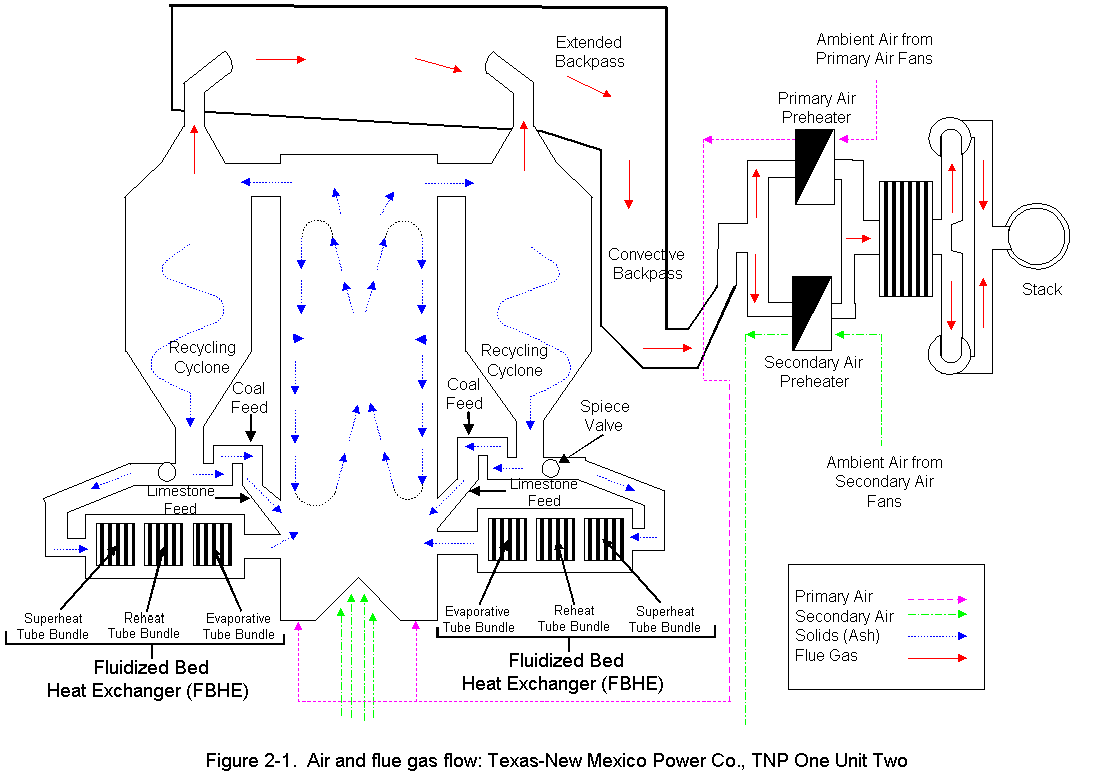{kind=link}
{kind=link}
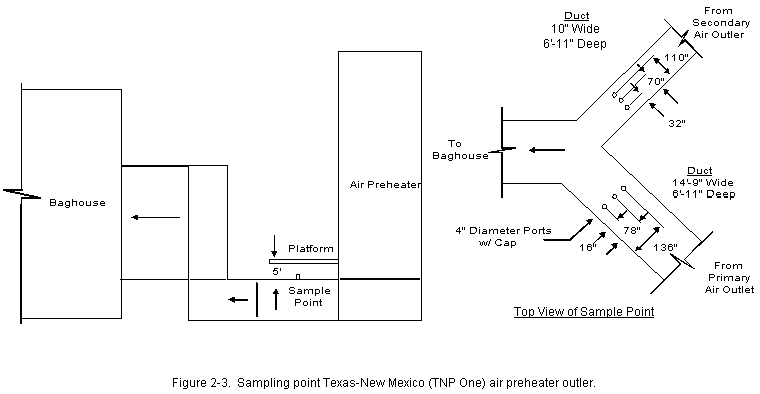{kind=link}
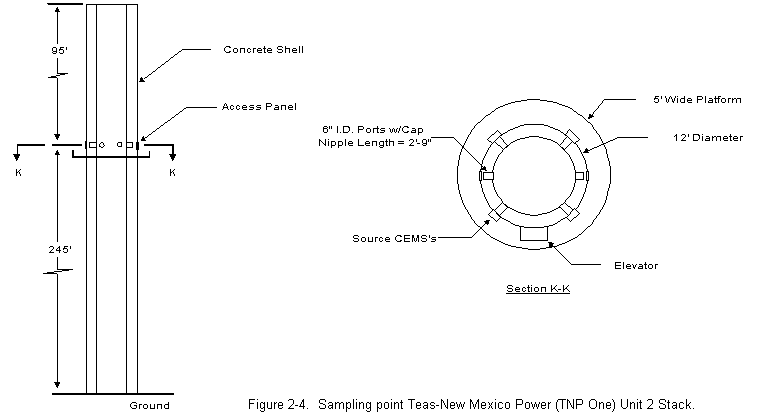{kind=link}
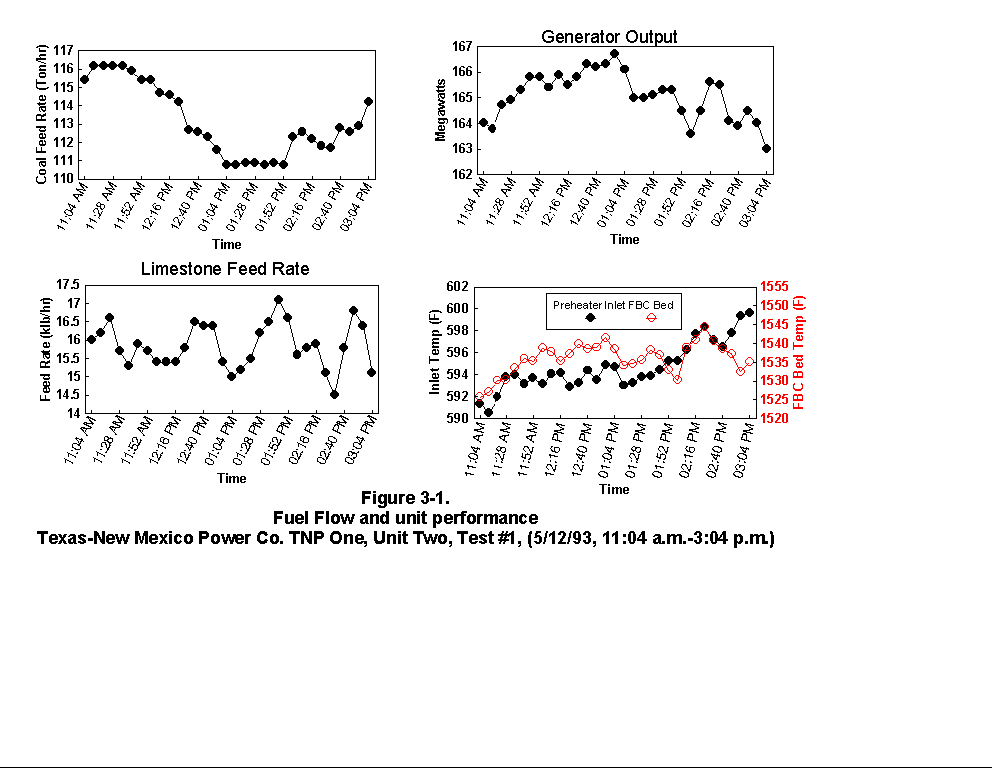{kind=link}
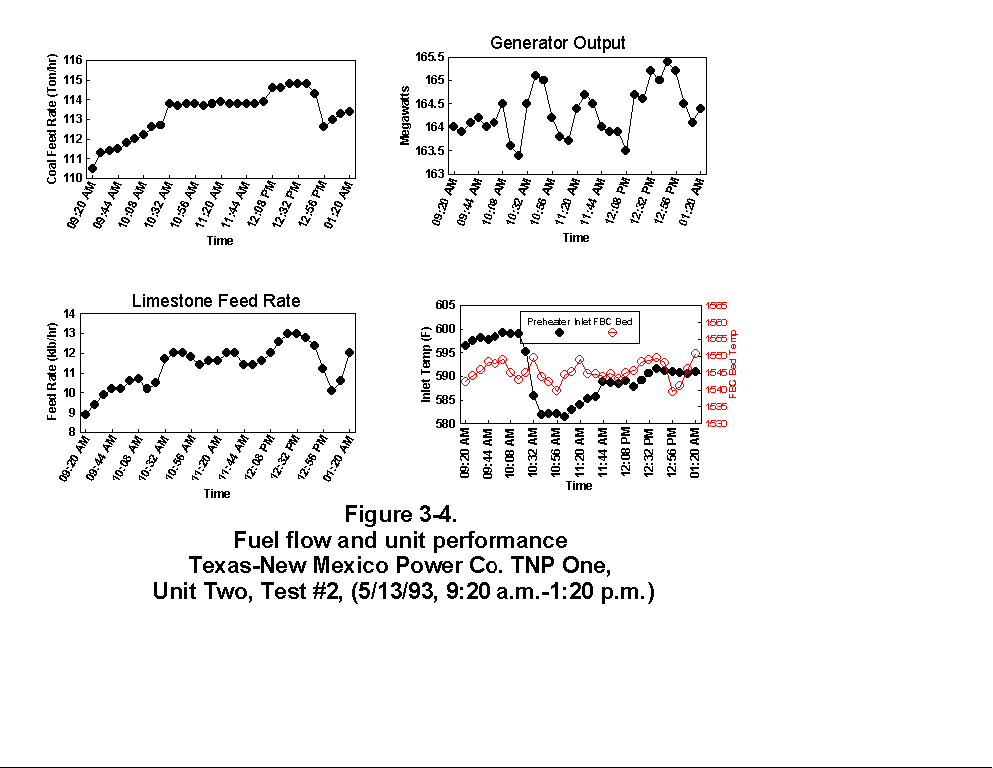{kind=link}
{kind=link}
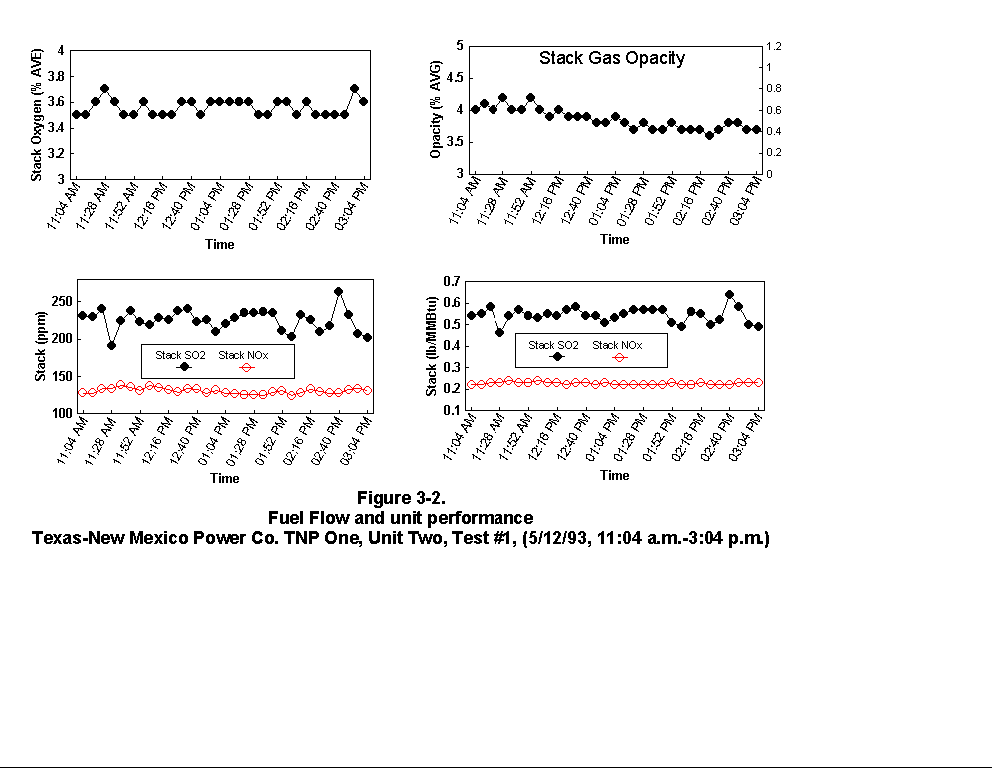{kind=link}
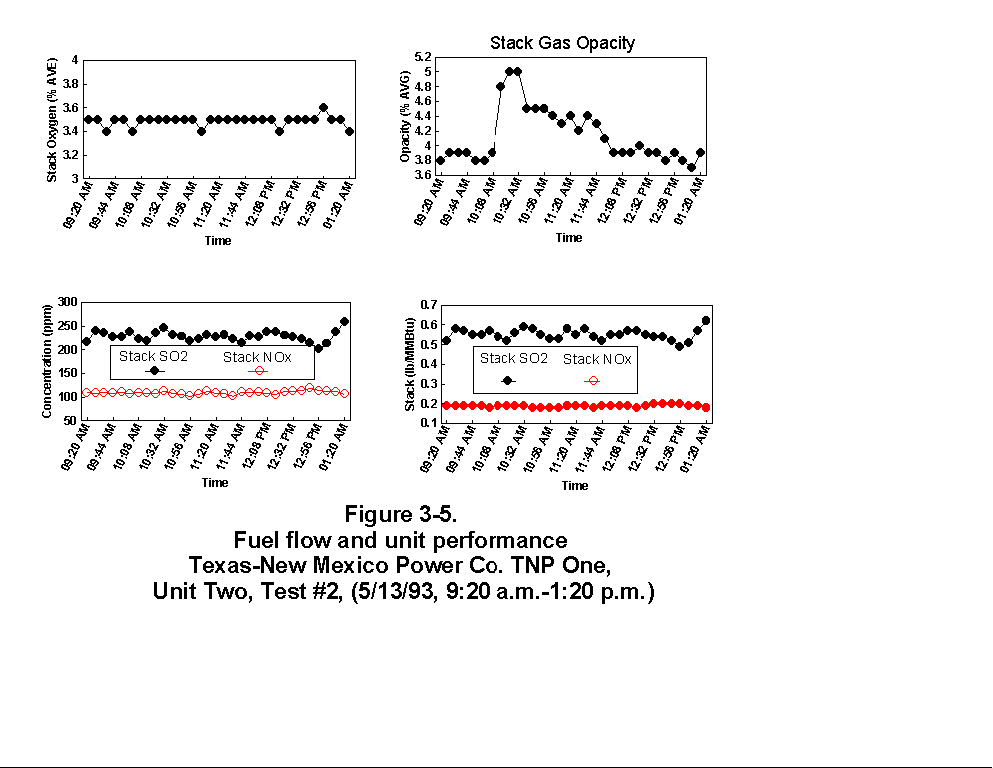{kind=link}
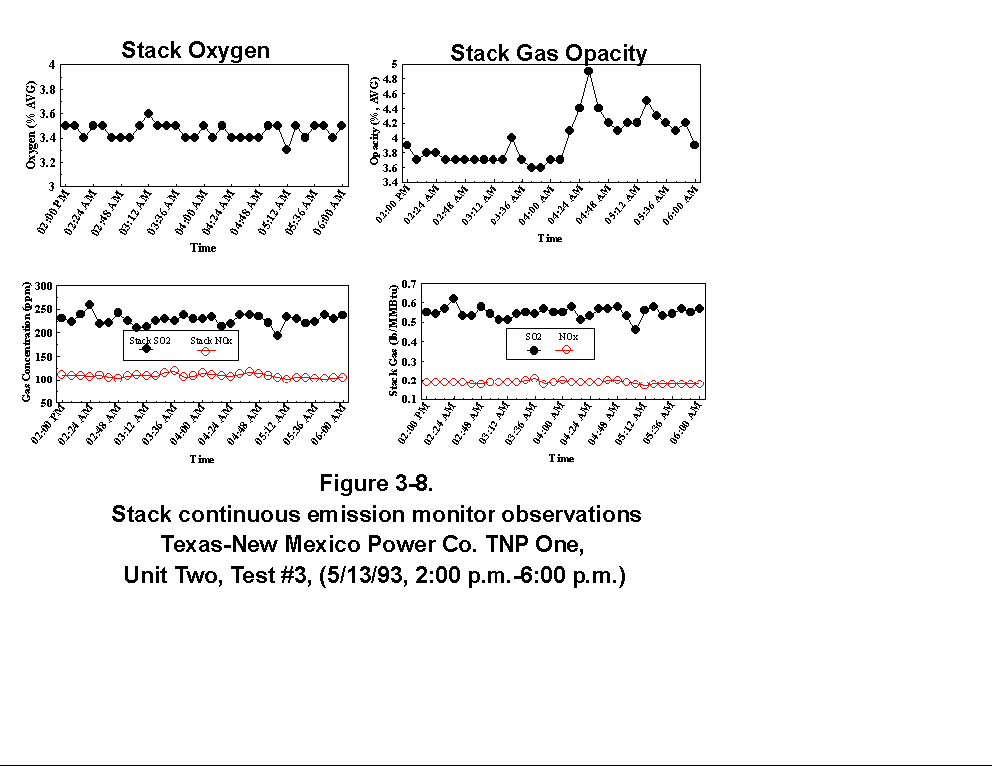{kind=link}
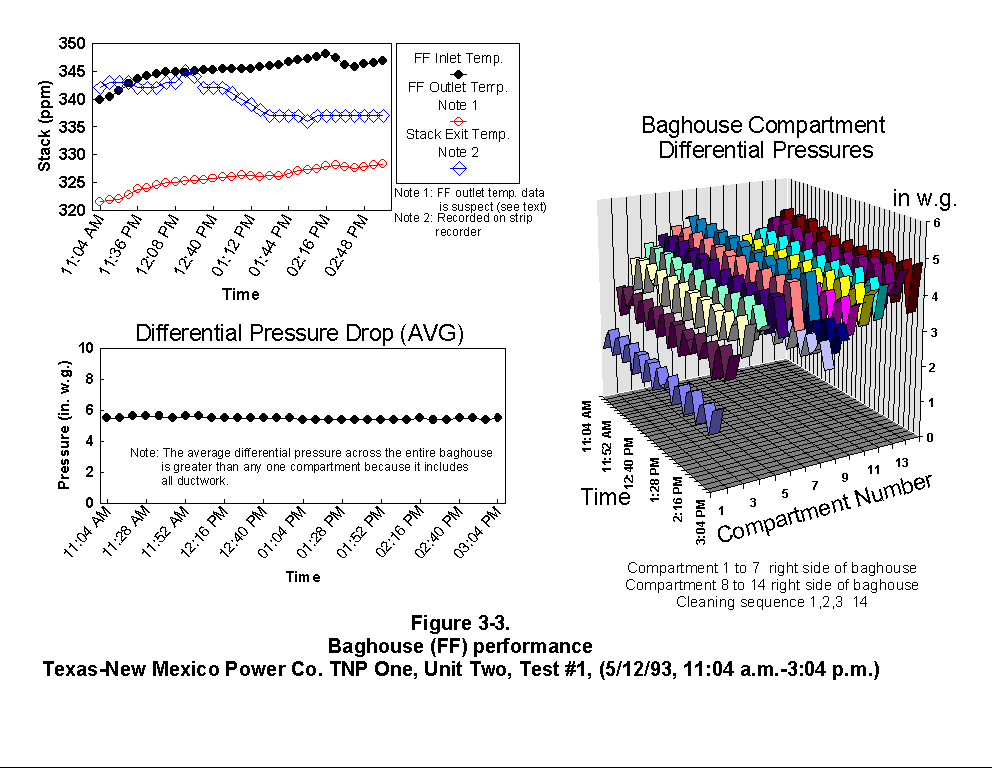{kind=link}
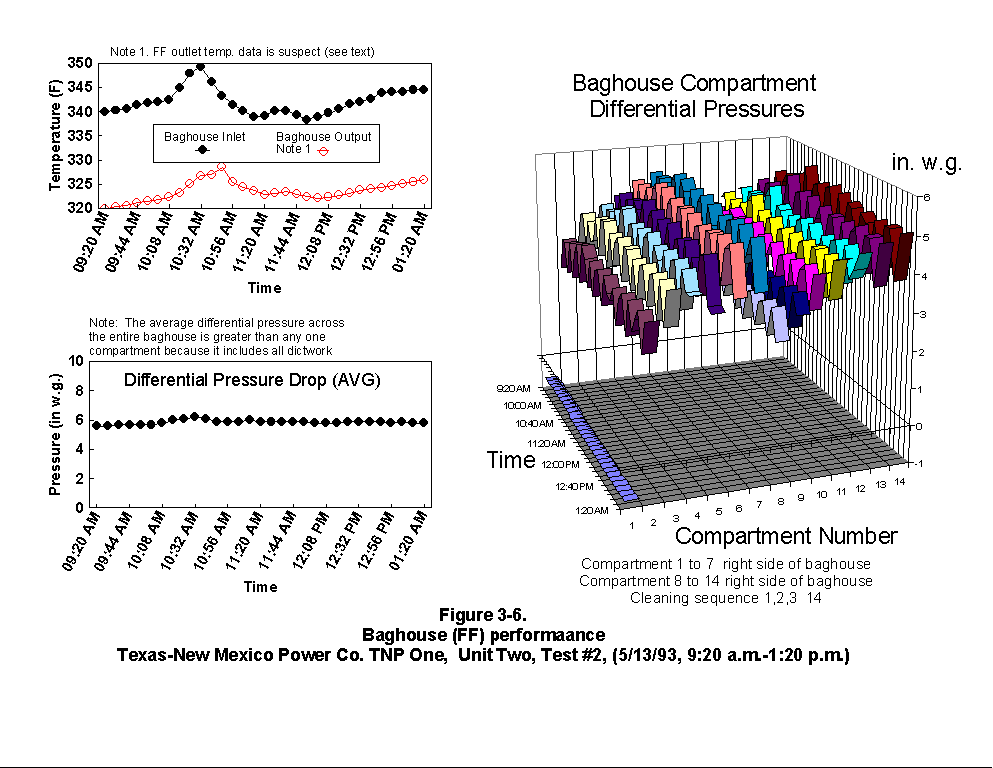{kind=link}
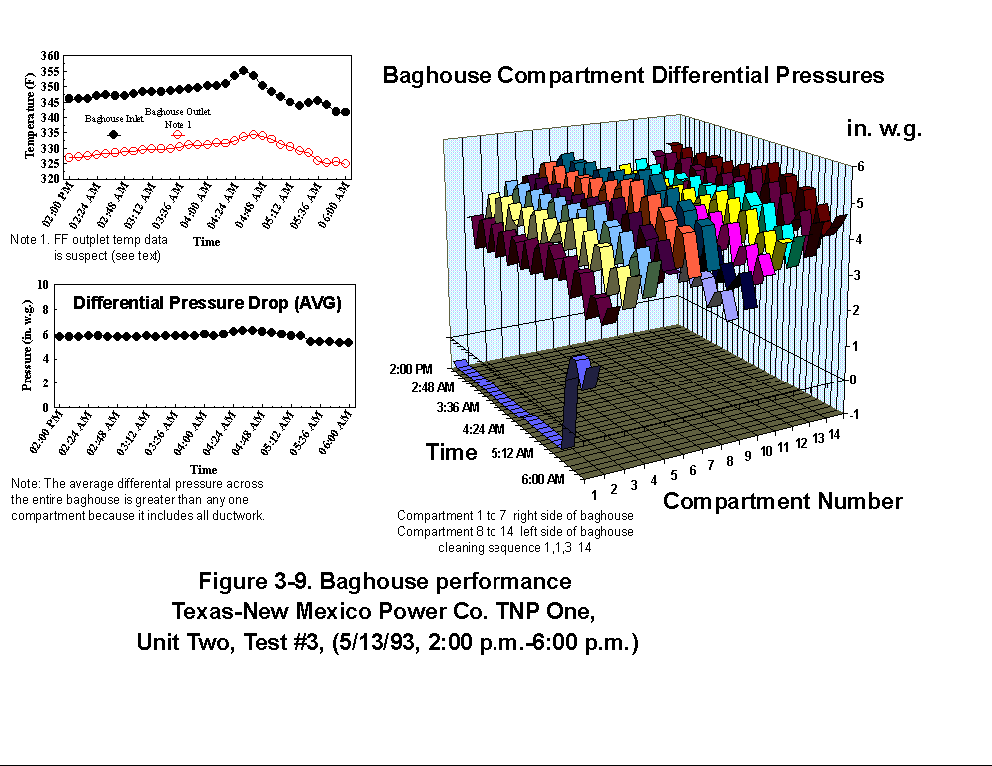{kind=link}
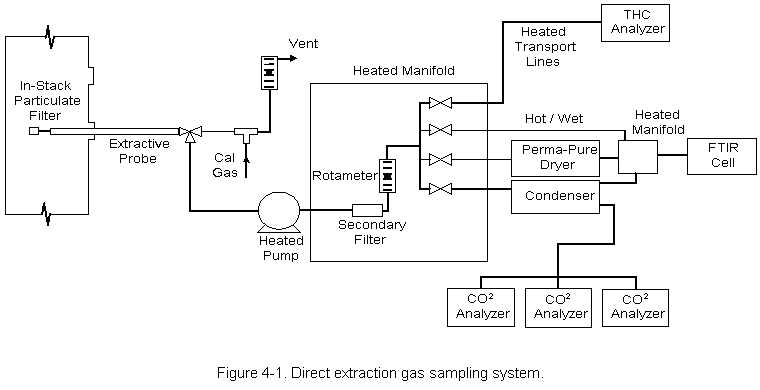{kind=link}
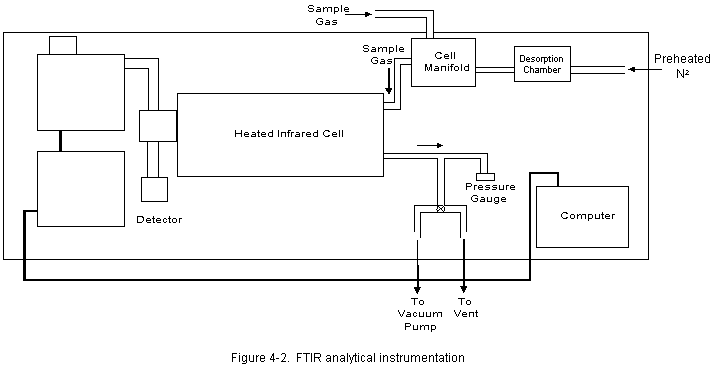{kind=link}
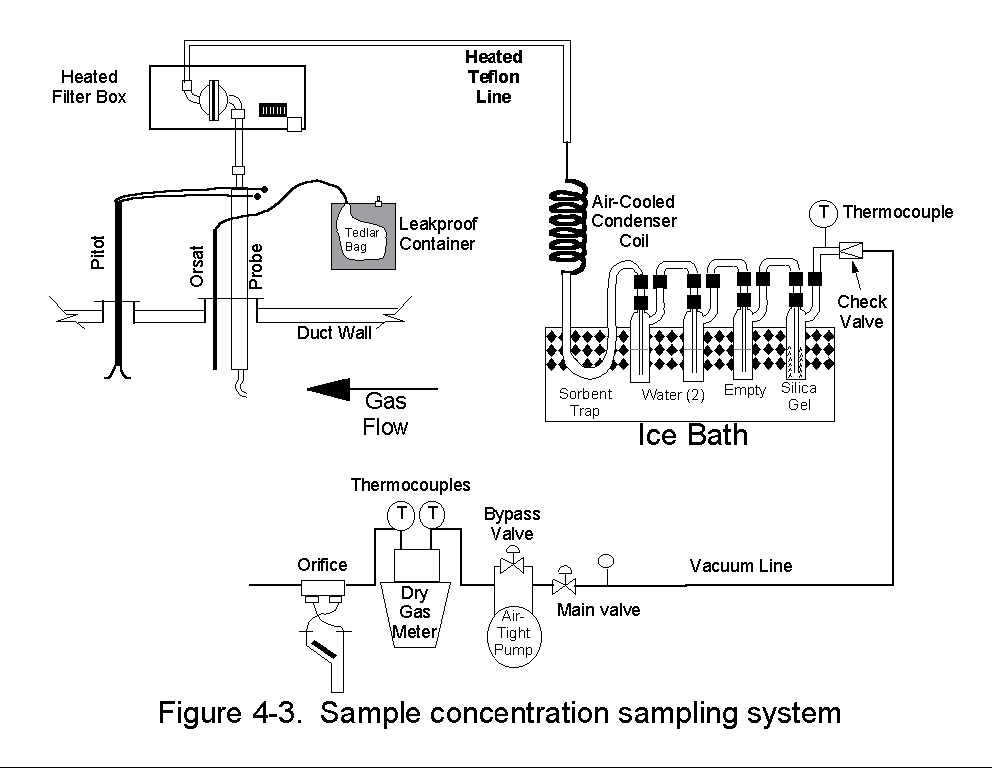{kind=link}